Pharmacodynamics of Antimicrobial Agents:
Time-Dependent vs. Concentration-Dependent Killing
Richard Quintiliani, M.D.
Pharmacokinetics deals with the movement of a drug from its administration site to the place of its pharmacologic activity and its elimination from the body. Factors affecting the movement (kinetics) and fate of a drug in the body are: (1) release from the dosage form; (2) absorption from the site of administration into the bloodstream; (3) distribution to various parts of the body, including the site of action and (4) rate of elimination from the body via metabolism or excretion of unchanged drug.
Although we cannot yet measure the drug concentration directly at the site of attachment to the bacterium, we can measure the drug levels in serum and other tissues as a function of time, thus using these surrogate levels to determine the concentrations of the antibiotic that are necessary to inhibit (MIC) or to be bactericidal MBC) to microorganisms. Drug concentration in the blood (plasma, serum) has been correlated to in vivo bacterial eradication. Most bacteria reside on the outside membranes of the cell, thus exposed to interstitial fluids. Drug concentrations in interstitial fluid drive the antibiotic into the bacterium and, ultimately, to the antibiotics binding site within the organism. Interstitial fluid drug concentrations are proportional to and in rapid equilibrium with blood and, therefore, the concentration of the antibiotic correlates with bacterial eradication.
By merely comparing the MIC or MBC’s of an antibiotic against a target organism, clinicians can draw the erroneous conclusion that the agent with the lowest MIC or MBC against a bacterium becomes the preferred choice. The MIC of an antibiotic against a pathogen is, however, only one of many factors that determine the best drug to cure an infection. When determining the potency of an antibiotic against a bacterium, other items such as protein binding, pharmacokinetics, distribution into the site of infection, the adequacy of the patient’s host defenses and the amount of exposure of an organism to an antibiotic needed for its eradication are also important considerations.
Pharmacodynamics correlates the concentration of the drug with its pharmacological or clinical effects. For an antibiotic, this correlation refers to the ability of the drug to kill or inhibit the growth of microorganisms. Antibiotics elicit their activity against bacterial by binding to a specific protein or structure in the organism.
For an antibiotic to eradicate an organism, three major factors must occur. First, the antibiotic must bind to its target site(s) in the bacterium. To reach the binding site is no easy matter. It must penetrate the outer membrane of the organism (penetration resistance), avoid being pumped out of the membrane (efflux pump resistance), and remain intact as a molecule (e.g., avoid hydrolysis by beta-lactamases). Once the target is reached, the antibiotic can still be useless if the binding site has changed its molecular configuration and no longer allows the drug to attach. A range of different binding sites has been identified including ribosomes, penicillin-binding proteins, DNA topoisomerase/gyrase, and the cell membrane itself. The crucial binding site will vary with the antibiotic class. These binding sites can be defined as points of biochemical reaction crucial to the survival of the bacterium. Thus, by binding to these sites, the antibiotic interferes with the chemical reaction resulting in the death of the bacterium.
Second, the drug must not only attach to its binding target but also must occupy an adequate number of binding sites, which is related to its concentration within the microorganism.
Third, for an antibiotic to work effectively, the antibiotic should remain at the binding site for a sufficient period of time in order for the metabolic processes of the bacteria to be sufficiently inhibited.
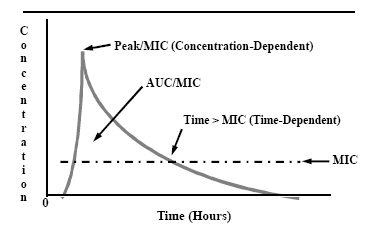
Thus, the two major determinants of bacteria killing include the concentration and the time that the antibiotic remains on these binding sites. The area under the serum concentration curve (AUC) after a dose of antibiotic measures how high (concentration) and how long (time) the antibiotic levels remain above the target MIC during any one dosing interval. In essence, the AUC indirectly measures the two major factors for bacterial eradication and quantifies the amount of exposure of the organism to the antibiotic during any one dosing interval.
Time-Dependent Killing:
For certain classes of antibiotics, the major killing effect against an organism is produced by either the time or the concentration of the drug at the binding site. In fact, of these two factors of bacterial killing, the killing process may be so minimal that it can be ignored in the prediction of a clinical response. For instance, certain antibiotics, like beta-lactams (penicillins, cephalosporins, carbapenems, monobactams), clindamycin, macrolides (erythromycin, clarithromycin), oxazolidinones (linezolid), can be effective because of the extensive amount of time the antibiotic binds to the microorganism. The inhibitory effect can be effective because their concentration exceeds the MIC for the microorganism. Hence, these antibiotics are referred to as time-dependent antibiotics. For time-dependent drugs, the pharmacodynamic parameter can be simplified to the time that serum concentrations remain above the MIC during the dosing interval (t>MIC) (Figure 1).
Concentration-Dependent Killing:
Other classes of antibiotics, such as aminoglycosides and quinolones, have high concentrations at the binding site which eradicates the microorganism and, hence, these drugs are considered to have a different kind of bacterial killing, named concentration-dependent killing. For concentration-dependent agents, the pharmacodynamic parameter can be simplified as a peak/MIC ratio (Figure 1).
These concepts have been even further refined from studies performed in animal models of sepsis, in-vitro pharmacokinetic models and volunteer studies. For instance, for antibiotics with time-dependent killing, the optimal responses occur when the time that the drug remains above the MIC is equal or greater than 50% of the dosing interval. For agents with concentration-dependent killing, the best responses occur when the concentrations are > 10 times above the MIC for their target organism (s) at the site of infection (1) For agents with concentration-dependent killing, it has also been shown that clinical responses can be predicted as well as the peak/MIC ratio by measuring the AUC over the dosing interval and dividing that value by the antibiotic’s MIC against the target organism. In essence, the AUC/MIC ratio becomes a “default” pharmacodynamic concept for the peak/MIC ratio for antibiotics with concentration-dependent killing.
This concept has been studied best with the fluoroquinolones. For instance, certain organisms require modest AUC/MIC ratio for their prompt eradication, Streptococcus pneumoniae and most other Gram-positive bacteria are typically rapidly killed by quinolones at an AUC/MIC24hr ratio > 30) whereas others, like Pseudomonas aeruginosa and most other aerobic Gram-negative bacteria, require much greater exposure of time to quinolones (AUC/MIC24hr ratios > 100-125) (2,3).
It must be emphasized that once these target AUC/MIC ratios, Peak/MIC ratio and tMIC are achieved there is no evidence that higher ratios result in more rapid killing or less emergence of bacterial resistance. In fact, excessive AUC/MIC ratios may produce unwanted adverse reactions by disrupting the normal gastrointestinal flora (“collateral damage”) and producing organ dysfunction.
REFERENCES
1. Craig W. Pharmacodynamics of antimicrobial agents as a basis for determining dosage regimens. Eur J Clin Microbiol Infect Dis 1993;12:Suppl 1:S6-8.
2. Dudley MN. Pharmacodynamics and pharmacokinetics of antibiotics with special reference to the fluoroquinolones. Am J Med 1991;91:45S-50S.
3. Zhanel GG, Walters M, Laing N, Hoban DJ. In vitro pharmacodynamic modeling simulating free serum concentrations of fluoroquinolones against multidrug-resistant Streptococcus pneumoniae. J Antimicrob Chemother 2001;47:435-440.
Recommended Reading:
McKinnon PS, Davis SL. Pharmacokinetic and pharmacodynamic issues in the treatment of bacterial infectious diseases. Eur J Clin Microbiol Infect Dis. 2004 Apr;23(4):271-88
Nuermberger E, Grosset J. Pharmacokinetic and pharmacodynamic issues in the treatment of mycobacterial infections. Eur J Clin Microbiol Infect Dis 2004;23:243-255.
Groll AH, Kolve H. Antifungal agents: in vitro susceptibility testing, pharmacodynamics, and prospects for combination therapy. Eur J Clin Microbiol Infect Dis 2004;23:256-270.
Edwards G, Krishna S. Pharmacokinetic and pharmacodynamic issues in the treatment of parasitic infections. Eur J Clin Microbiol Infect Dis 2004;23:233-242.
Figure 1. Pharmacokinetic/Pharmacodynamic Parameters Affecting Antibiotic Potency